We have applied some changes to ARIA and are monitoring stability, but you can continue to work as normal. We appreciate your understanding.
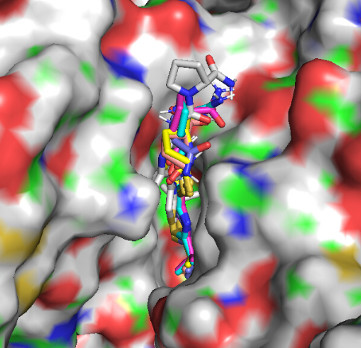
The Scipion-Chem Service enables researchers to transform structural information about biological macromolecules into actionable hypotheses for ligand binding and therapeutic development. The service provides a fully integrated environment for binding pocket identification, ligand docking, and virtual screening, allowing users to start with experimentally determined or modeled 3D structures and rapidly generate ranked lists of potential ligands. Users can submit their structures and (optionally) their own compound libraries or access curated public collections to explore new binding sites, prioritise hits, and guide experimental validation.
The service requires as input a 3D atomic structure of the target macromolecule. The user may optionally provide a library of small molecules in SMILES format; otherwise, the screening will be performed against a curated set of ~1500 FDA-approved compounds.
Scipion-Chem offers two main types of analyses:
Results are fully integrated within the Scipion environment, providing interactive 3D visualization of druggable sites, binding poses, and ranked compound lists. The outputs can be exported for further refinement, such as molecular dynamics simulations or medicinal chemistry optimization.
This service enables Instruct users to derive early insights into the druggability of newly solved macromolecular structures and to generate experimentally testable hypotheses about potential ligands. The integration into Scipion ensures traceability, reproducibility, and interoperability with other Instruct pipelines, including those for cryo-EM data processing and atomic model refinement.
Execution of a pocket finding or virtual drug screening workflow
Address: c/Darwin, 3, 28049, Madrid, Spain
Facility weblink: https://i2pc.es/
Physical access: yes
Remote access: yes
Average time of a visit: 4 days
Information about sample preparation: The service requires as input a 3D atomic structure of the target macromolecule